







This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Clinical Case - Test Yourself
A 6-year-old boy was administered to our Radiology Department for Magnetic Resonance Imaging (MRI) of two soft tissue tumours on the dorsal surface and on the distal lateral side of his right second toe. The nodules appeared skin-coloured and were quite firm but not painful on palpation (
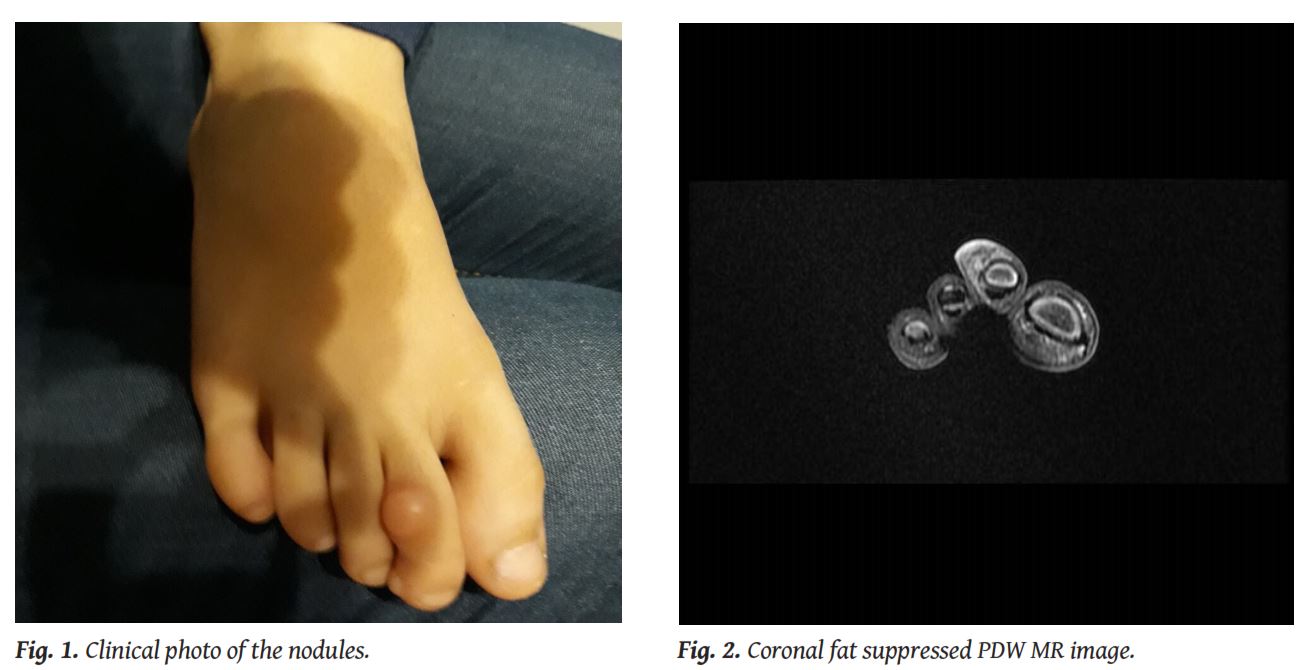
Infantile digital fibroma is a rare, relatively benign tumour, first described by Reye in 1965, with almost 200 cases reported in the literature so far. It appears as a pinkish or skin-coloured, broad-based nodule on the dorsal or lateral surface of the digits of both hands and feet, but usually sparing the thumb and the big toe [1]. Fingers have been found to be affected more commonly than the toes (60% vs 40% respectively) [1, 2]. The lesion can present as a solitary nodule, although simultaneous appearance of more than one lesions or even metachronous lesions have also been reported [1].
The main rule is that digital fibroma is either present at birth or, more typically, appears during the first three years of life, with reports for much later onset existing in a few sparse cases [1, 3]. The lesion shows a slight male predominance and its size ranges between 3 and 35 mm (usually up to 20 mm) [2].
At initial presentation, the tumour seems to grow slowly, followed by a several-months period of rapid growth, at the end of which the lesion remains rather stable [1]. Clinically, most of the lesions are asymptomatic, but if they become large enough, they can cause joint pain or functional and cosmetic problems. Digital fibromas can spontaneously involute. They do not metastasise, but they show a high rate of recurrence after surgical resection (almost 63%) [1-3].
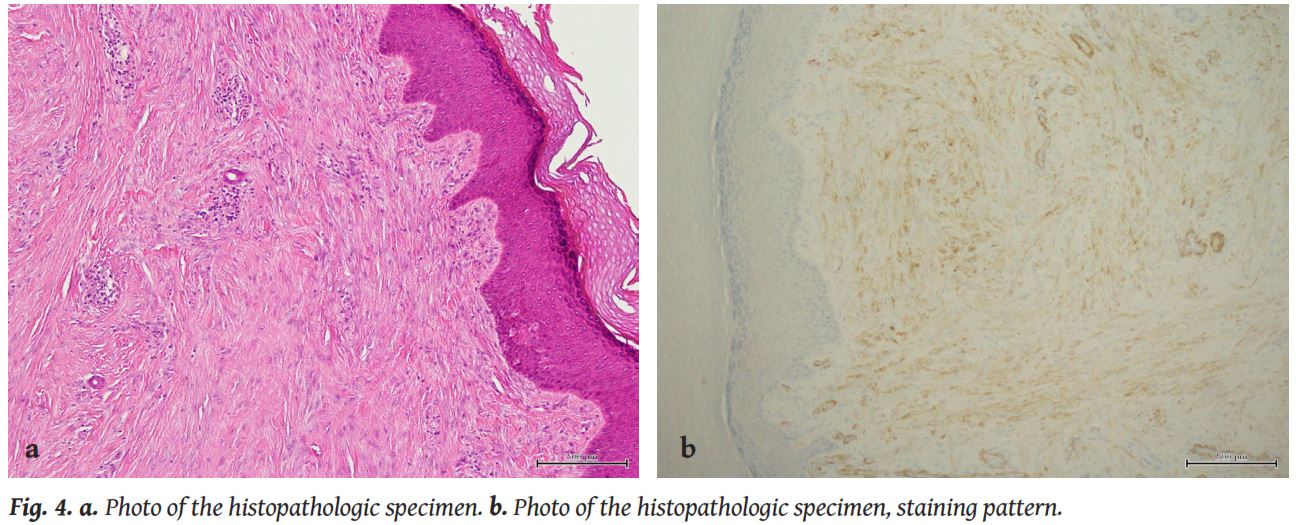
Pathologically, these tumours have a unique pathognomonic feature that helps distinguish them from other similar entities: the presence of perinuclear, intracytoplasmic inclusion bodies, that appear similar to red blood cells and are made of densely packed, vimentin and actin filaments [2, 4]. These inclusion bodies vary a lot in number. Early in their course, the lesions are less fibrotic and they harbour less intracytoplasmic bodies [2].
This unique histopathologic appearance of this entity, in addition to a few number of cases described in older populations, has led many researchers to suggest the implementation of strictly descriptive terms for the condition, such as “inclusion body myofibroblastoma” [4]. The term “infantile digital fibroma” is preferred for cases appearing during infancy.
The intracytoplasmic inclusion bodies, although pathognomonic for the establishment of the diagnosis, are not always present. Other important histopathologic features are then helpful to confirm the diagnosis, such as cellular proliferation in the dermis, collagen and spindle-shaped myofibroblasts in perpendicular fascicles beneath the skin and tumour cells that infiltrate the periadnexal adipose tissue of the dermis. Positive staining patterns forα-smooth muscle actin and desmin contribute to the diagnosis as well [5].
The aetiology and pathogenesis of fibromas are not fully known, but no genetic abnormality or syndrome has been recognised as relevant to their appearance [2].
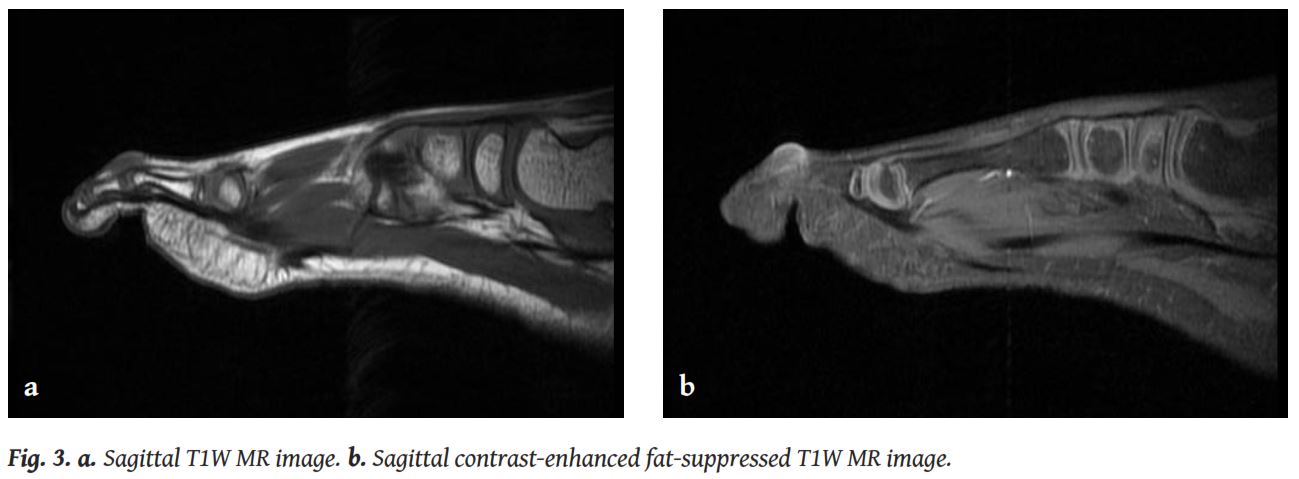
Digital fibromas are not a common entity to deal with in a Radiology Department. This is why the reports of imaging findings of this condition are very rare in the literature. Plain radiographs are expected to show a non-specific soft tissue swelling in the digit, with normal appearance of the adjacent bone. MRI of the lesion usually demonstrates a soft tissue nodule, with variable degree of enhancement, without involvement of the underlying bone. Indeed, there is only one case in the literature where invasion of the bone was observed [1].
The differential diagnosis includes infantile fibromatosis, infantile lipofibromatosis, calcifying aponeurotic fibroma, pachydermodactyly, keloids, terminal osseous dysplasia, pigmentary defects and palmar-plantar fibromatosis [2, 5].
Because of the high rates of local recurrence and the potential for spontaneous regression, the most accepted approach of the lesion is biopsy for the histologic confirmation, followed by firm clinical observation. Intralesional injections of triamcinolone, imiquimod and 5-fluorouracil have been occasionally applied as non-operative treatment of disabling cases [5]. Surgical treatment should be reserved for cases where the tumour shows increase in size on follow-ups [2].
1. Sargar KM, Sheybani EF, Shenoy A, et al. Pediatric fibroblastic and myofibroblastic tumors: a pictorial review.RadioGraphics2016; 36: 1195-1214.
2. Marks A, Ewart E. Infantile digital fibroma, a rare fibromatosis.Arch Pathol Lab Med2016; 140(10): 1153-1156.
3. Robbin MR, Murphey MD, Temple HT, et al. Imaging of musculoskeletal fibromatosis.RadioGraphics2001; 21(3): 585-600.
4. Mortimer G, Gibson AA. Recurring digital fibroma.J Clin Pathol1982; 35(8): 849-854.
5. Eypper EH, Lee JC, Tarasen AJ, et al. An algorithmic approach to the management of infantile digitalfibromatosis: Review of the literature and a case report.Eplasty2018; 18: e19.
None